患者男,68岁,因“渐进性四肢无力伴肌肉萎缩5年,加重1 年”入院。
现病史
患者自2003年起逐渐感到双下肢无力,主要表现为双下肢下蹲后起立困难,步行及上楼尚可。同时感到双手抓握困难。此后症状渐进性加重,并发现双大腿逐渐变细。近1年来双
下肢不能下蹲,坐在凳子上站立不起,需旁人帮助。近4个月来右腿外侧肌肉酸痛明显,左手抓握无力加重。于外院接受激素治疗症状无改善。病程中无吞咽困难,无感觉障碍,
精神可,胃纳和睡眠可,大小便如常。
既往有风湿性心脏病史及扁桃体摘除史。否认家族遗传病史。
体格检查和辅助检查
查体:神志清楚、对答切题。双侧瞳孔等大等圆,对光反应灵敏,视野无缺损,眼球各向活动佳。鼻唇沟对称,伸舌居中。颈软,抬头肌力4级。左上肢肌力
肩5级-肘4级腕4级-指3级,右上肢肌力肩5级-肘4级腕4级指4级。双手屈指困难,左侧为甚。左下肢肌力髋4级-膝4级-踝5级-趾5级-,右下肢肌力髋4级膝4级踝5级-趾5级-。双侧
膝、踝反射减弱。双侧病理征(-)。双侧大腿近端股四头肌萎缩明显,左侧较明显。全身深浅感觉无明显异常。共济检查正常。
实验室检查:
血常规:白细胞计数 5.89 × 10E9/L(参考值:4.5~11.0 × 10E9/L),红细胞计数 4.35 × 10E12/L(参考值:4~5.50 × 10E12/L),血红蛋白 136 g·L-1(参考值
:120~160 g/L),中性粒细胞67.6%(参考值:45.0~70.0%),血小板计数 119×10E9/L (参考值:100~300×10E9/L)。
肝功能:丙氨酸转氨酶 36 U/L(参考值:<50 U/L),天冬氨酸转氨酶 26 U/L(参考值:<30 U/L),总胆红素 20mmol/L(参考值:3.4~20.4 mmol/L),结合胆红素 6.3mmol/L(参考值
:<6.8 mmol/L),总胆汁酸9 mmol/L(参考值:<10 mmol/L),碱性磷酸酶 51 U/L(参考值:40~129 U/L)。
肌酶:肌酸激酶CK 400 U/L(参考值:38~174 U/L),肌酸激酶同功酶 21 U/L(参考值:<25U/L),a-羟丁酸脱氢酶 153 U/L(参考值:72~182 U/L),乳酸脱氢酶 188 U/L(参考值:
106~211 U/L)。
血电解质:钾3.6 mmol/L(参考值:3.5~5.5 mmol/L),钠144mmol/L(参考值:135~147 mmol/L),氯化物 106mmol/L(参考值:95~105 mmol/L),钙 2.24 mmol/L(参考值:
2.10~2.60 mmol/L),镁0.93 mmol/L(参考值:0.60~1.10 mmol/L)。肾功能:尿素氮5.0 mmol/L(参考值:2.5~7.0 mmol/L),血肌酐60 mmol/L(参考值:50~80 mmol/L),尿酸
0.340 mmol/L(参考值:0.100~0.420 mmol/L)。
空腹血糖:4.8 mmol/L(参考值:3.9~5.8mmol/L)。凝血功能:国际标准化比率:1.11(参考值:0.91~1.10),凝血酶原时间13.1(参考值:10.8~12.8),活化全血凝固时间34.1 s(
参考值:21.1~36.5 s),纤维蛋白原 1.9 g·L-1(参考值:1.8~3.5 g/L)。
肿瘤标志物:CA125(cancer antigen)<35.00 U/L(参考值:<35.00 U/L),CA199<37.00 U/mL(参考值:<35.00 U/mL),CA724 1.86U/mL(参考值:<8.20 U/L), PSA(prostate
specificantigen) 2.60 ng/L(参考值:4.10 ng/L),FPSA(free prostate specific antigen) 0.76 ng/L,FPSA/PSA 0.29(参考值:>0.25,当PSA>4.1 时才有判别价值),AFP
(alpha fetoprotein)1.74 mg/L(参考值:10.00 mg/L),CEA(carcino embryonic antigen) <0.64 mg/L(参考值:10.00mg/L)。
肌电图(EMG):部分被检肌静息下见正尖、纤颤波(左侧桡侧腕屈肌、肱二头肌,右侧胫前肌和股内肌)。收缩MUP见短棘多相波和不规则波;右侧腓内肌轻收缩AMUP偏宽大;右股内
肌重收缩募集呈单纯相。被检感觉A和运动神经传导速度和波幅正常范围(正中神经、尺神经、腓总神经和腓浅神经)。提示为活动性肌源性损害合并轻度神经源性肌电改变,以肌源
性损害为主。
请问:该例患者肌无力及萎缩原因考虑何种肌病所致?
请大家依据所给资料进行诊断,诊断正确或诊断思路清晰、有理有据者会酌情给予积分奖励!
答案及讨论将于一周后(2012-10-22)公布!
公布答案:包涵体肌炎
诊断分析:
本病例表现为进行性四肢不对称性肌无力及肌肉萎缩,近端和远端均有累及。上肢以远端为主,屈指肌力减退重于伸指肌力。双下肢以近端股四头肌为主,肌肉
无力和萎缩左右不对称,左侧股四头肌萎缩更显著。不伴有感觉症状,无肉跳。查体有膝踝反射减弱。故可定位于下运动神经元或肌肉。患者的临床特点以双下肢近
端不对称性的肌无力和肌肉萎缩为主要表现,并且有累及双上肢远端肌肉。
临床上表现为不对称的双下肢近端肌无力的疾病见表1。

包涵体肌炎(inclusion body myositis,IBM) 是一种常见于50 岁以上的炎性肌病。男女比例为3:1,缓慢隐袭起病,主要表现为上肢远端和下肢近端肌肉无力和萎缩,具有不对称性和选择性,以指深屈肌和股四头肌受累最明显。患者可以出现持续性的屈腕、屈指和伸膝无力。肌酶升高不超过正常值的12 倍。EMG表现为肌
源性损害为主,有时可合并神经源性损害。肌肉在电镜下见到肌浆和肌核内15~18 nm 的管状细丝包涵体是诊断依据。
局灶性肌炎(local myositis) 多发生于30~50岁的成年人。男女发病比例大致相等。临床表现为肌无力和肌痛。可仅累及单个肌群,或不对称累及双侧,多见于
股四头肌受累,也可有腹部、上臂、前臂、颈部、舌肌以及口周肌肉的累及。血清肌酸激酶多为正常。
肢带型肌营养不良2B型(limb-girdle muscular dystrophy2B,LGMD 2B) 为常染色体隐性遗传的先天性肌营养不良,突变位于染色体2p13.3-p13.1。20 岁左
右发病,进展缓慢。临床上以下肢近端肌无力起病,股四头肌和腰肌最易受累。肌肉无力和萎缩常不对称。晚期可累及上肢近端,但远端手肌不受累。实验室检查血
清肌酸激酶显著升高,为正常值的10~72倍。EMG表现为肌源性损害。肌肉病理免疫染色可见肌膜Dysferlin染色缺失或减少,肌浆染色缺如。确诊需依赖蛋白免疫印迹或DNA分析结果。
McArdle病(phosphorylase deficiency,MrArdle’s)糖原累积病Ⅴ型,又名肌肉磷酸化酶缺乏症。为常染色体隐性遗传病,主要突变在11q13。通常在15岁以下起病,也有50岁以上发病。男性发病为主。临床上主要表现为长时间剧烈运动或劳动后出现肌肉酸痛和抽搐。肌肉无力表现多样,四肢近端受累为主,远端手部也可无力,常不对称,休息后肌肉酸痛好转。约有12%的患者会出现肌肉萎缩,类似于本病例。但有9%的患者伴有腓肠肌肥大。实验室检查血清肌酸激酶静息状态下仍显著升高。EMG提示肌源性损害。前臂缺血试验乳酸增高,但不超过正常值3 倍,但确诊仍需酶生化检查。
糖尿病性肌萎缩(diabetic amyotrophy) 发生在糖尿病患者的肌肉无力和萎缩。老年男性多见,且糖尿病Ⅱ型比Ⅰ型更容易发生。亚急性起病。病损累及腰骶神经丛和神经根,临床表现为一侧或两侧下肢近端肌肉无力和萎缩,股四头肌最易受累,伴有疼痛或膝反射消失。少数累及上肢无力。其诊断主要依靠电生理检查,EMG可见到棘旁和腿部肌肉多灶性失神经支配和轴突损害,提示腰骶神经根、神经丛或股神经的神经源性损害。腰椎穿刺脑脊液检查蛋白明显增高,细胞数正常。血清肌酶正常范围。
脊髓灰质炎后综合征(postpoliomyelitis syndrome)为脊髓灰质炎患者病后有肢体完全或部分单肢或多肢体瘫痪。病后15年又重新出现原瘫痪和以前从未瘫痪的肌群乏力、肌痛、疲劳和萎缩的一种综合征。无力和萎缩多为不对称性,双手和双下肢受累较常见。病程进展缓慢。EMG提示脊髓前角细胞损害。有脊髓灰质炎病史和其造成的肢体瘫痪,病后15年余又出现原瘫痪肢体或未累及肢体的下运动神经元损害是诊断的主要依据。
最后诊断及分析:
该患者63 岁起病,起病隐匿,进行性加重。以双下肢近端肌无力起病,左右不对称,以后出现双上肢远端无力,屈指无力和伸膝无力明显。实验室检查血清肌酸激酶轻度升高。EMG提示肌源性损害为主,合并轻度神经源性损害。患者无明显家族遗传病史,对激素治疗无明显效果,故考虑IBM可能。
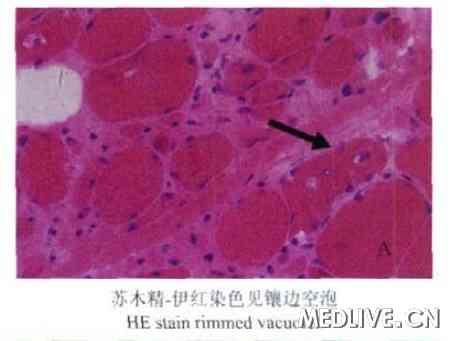
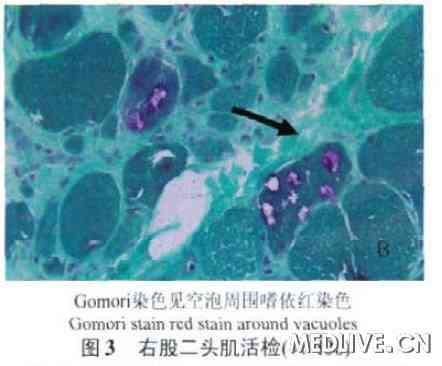
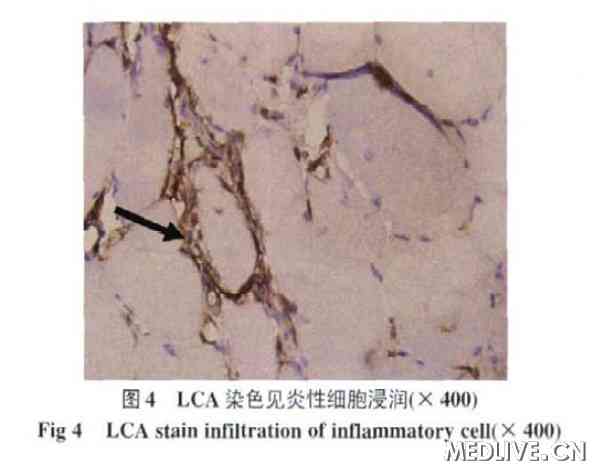
行右股二头肌活检,苏木精- 伊红染色镜下可见肌纤维明显大小不一,形态极不规则,肌周核显著增多,核内移明显增多(>3%)。部分肌纤维萎缩、变性。部分肌纤维内可见镶边空泡(图2),Gomori染色镶边空泡更为显著(图3)。LCA (leukocyte cammon antigen)染色可见明显的炎性细胞浸润(图4)。
ATPase 染色见Ⅰ、Ⅱ型肌纤维镶嵌排列。SDH(succinate dehydrogenase)、NADH(nicotinamide adenine dinucleotide)及PAS(PolysulfonamidePoly sulfide polyaryl)染色,油红O 染色未见明显异常,
COX(cyclooxygenase)染色缺失。
免疫组化分析:肌纤维肌细胞膜肌萎缩蛋白等未见明显缺失。细胞LCA(+),KP-1(+)。电镜下可见部分细胞肌丝溶解、融合、紊乱稀疏排
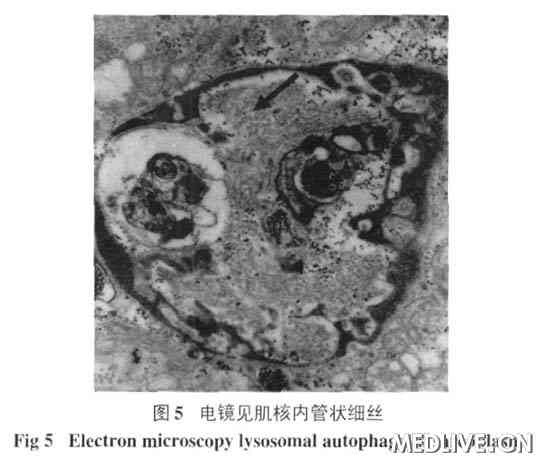
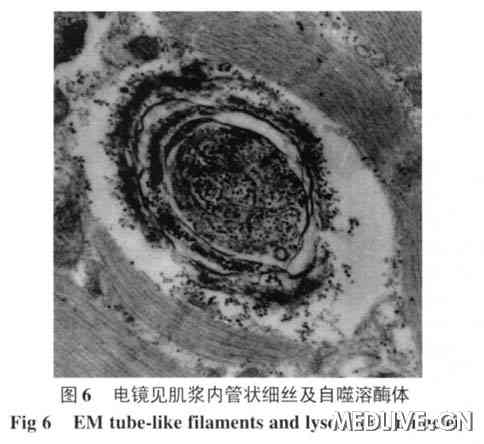
列,Z线缺失,线粒体肿涨、嵴脱失。肌质网无扩张。糖原略多。有较多髓样体和自噬溶酶体。在光镜下所见的8 个镶边空泡,其中有2 个可在电镜下见到肌浆或肌核
内管状细丝包涵体(图5,6)。
结合肌活检,发现典型的镶边空泡,肌膜部分增生,有炎性细胞浸润。电镜下见肌核和肌浆内管状细丝包涵体及髓样物质。综合病史、临床表现和病理学特点,IBM诊断明确。
IBM被认为是一种好发于成年人的慢性进行性骨骼肌炎性疾病。1960 年,由Chou 首先报道,Yunio 等于1971 年首次提出IBM 这一疾病名称。但直到1978 年Carpenter对14例IBM的临床病理特点进行了总结后,才正式确立了IBM为一独立的疾病。
该病多在50 岁以后发病,男性多见,起病隐匿,病程进展缓慢。典型表现为四肢肌肉无力和肌肉萎缩,远端和近端均有累及,具有选择性和不对称性。约70%的首发症状为下肢近端无力,以股四头肌受累最常见。也可以下肢远端、上肢或四肢无力起病,常为非对称分布。
10% 的患者早期出现上肢远端无力,指深屈肌受累较多。40%~80%的患者晚期出现吞咽困难。少数患者累及面肌。肌无力进行性加重,以屈指、屈腕及伸膝肌力减弱最显著。腱反射常减低,尤以膝反射减弱最为常见。少数患者可有感觉异常。
IBM常可合并其他疾病,按其发生率依次为心血管疾病、周围神经病、糖尿病、自身免
疫性疾病(间质性肺炎、银屑病、红斑狼疮和皮肌炎)。
本例患者合并有风湿性心脏病和扁桃体炎,但这些并发症与包涵体肌炎之间的关系目前尚不能确定。
实验室检查中多数患者的血清肌酸激酶水平正常或轻度升高,一般不超过正常值上限的12 倍。EMG主要呈肌源性损害,亦可见神经源性损害,但运动、感觉神经传导速度正常。IBM的EMG损害表现主要有3型:①纤颤电位/正向尖波和短时程/小振幅的MUP占56.6%;②纤颤电位/正向尖波、长时程/高振幅的MUP占36.7%;③纤颤电位/ 正向尖波、正常和长时程/ 高振幅MUP占6.7%。本例患者的EMG主要表现为纤颤正尖波和短时程/小振幅的MUP。
肌活检是目前诊断的主要检查方法。肌肉病理可见炎性细胞浸润肌内膜,肌纤维内出现异常结构和蛋白。镶边空泡是IBM的病理学特点之一,在空泡内常可见嗜酸性胞浆体和类淀粉物质。空泡以股四头肌最常见,肱二头肌次之,三角肌常无。电镜检查发现肌浆或肌核内管状细丝包涵体是本病重要的病理学特征,但电镜下寻找包涵体不容易,本病例选择了光镜下的8个空泡纤维,发现两个包涵体(图5、6),这种包涵体由盘绕的管状细丝组成,细丝外径10~20 nm,内径3.6~8 nm,长为1~5mm。肌浆内的包涵体可能来自于肌核,细丝可以呈相互平行或向心性排列,也可以杂乱无序。其周边常包绕糖原颗粒、不规则的髓样结构、膜碎片及胞质的分解产物。如同图5中管状细丝周边包绕着较多髓样体及糖原颗粒。部分患者的骨骼肌可以出现破碎红细胞纤维RRF(ragged red fibers)和细胞色素C氧化酶阴性肌纤维以及肌病样改变或神经源性改变。
关于IBM的确切病因和发病机制至今未明,但是近来认为自身免疫和变性两个机制均有可能。最早报道包涵体的时候,曾怀疑其为一黏病毒产物,后来又发现
麻疹病毒抗体能与IBM的包涵体结合,但患者肌纤维中均找不到病毒颗粒,应用PCR扩增均得到阴性结果。尽管如此,仍有12个病例报道IBM与逆转录病毒感染相关,故提示慢性持续的病毒感染可能是潜在的诱发因素。
IBM变性发病机制的依据有:主要是由于IBM的分子病理表现与阿尔茨海默病(Alzheimer’s disease,AD)和帕金森病(Parkinson’s disease)等最常见的神经变性疾病的脑组织分子病理表现非常相似,两者都有以下成分的聚集:β1淀粉样蛋白前体(β-APP)、β淀粉样蛋白、在成对螺旋丝(PHF)形成中含有磷酸化的tau 蛋白、早老素(presenilin-1)以及其他几种AD的特异性蛋白如载脂蛋白E(apoE)和泛素。IBM空泡样肌纤维中β-APP mRNA亦增高,还可见乙酰胆碱受体(AChR)、朊蛋白和其mRNA蓄积。
本病目前缺乏特效治疗。类固醇激素能降低血清CK水平,但镶边空泡纤维和嗜刚果红物质均增加,临床肌无力加重或仅有轻微改善,这是区别于皮肌炎/多发性
肌炎(PM/DM)的重要临床特征。也可补充提示IBM的炎症反应可能为继发性的。静脉注射免疫球蛋白可改善部分患者的无力,但程度有限,是否有效还需进一步临
床试验来证实。IBM应用免疫抑制剂后可稍有改善,但总体预后不佳,其治疗效果远远差于多发性肌炎。近年来对于IBM的治疗研究认为抗淋巴细胞信号传导的单克隆抗体和抗细胞因子治疗可能阻止疾病的进展。患者一般在发病15年后生活不能自理,常因吞咽困难和呼吸肌瘫痪死亡。
病例来源:中国临床神经科学 2009 年第 17卷第4期
作者:复旦大学附属华山医院
黄 莹,赵重波,卢家红,奚剑英,俞 彰



 微信公众号
微信公众号